Projevy argininémie se často podobají jiným symptomům neurologických či neurometabolických poruch, mezi něž patří poruchy močovinového cyklu (UCD), mozková obrna (CP) či dědičná spastická paraplegie (HSP) 5,6.
Diferenční diagnóza ARG1-D zahrnuje kombinaci klinických projevů a zvýšenou koncentraci argininu4-7.
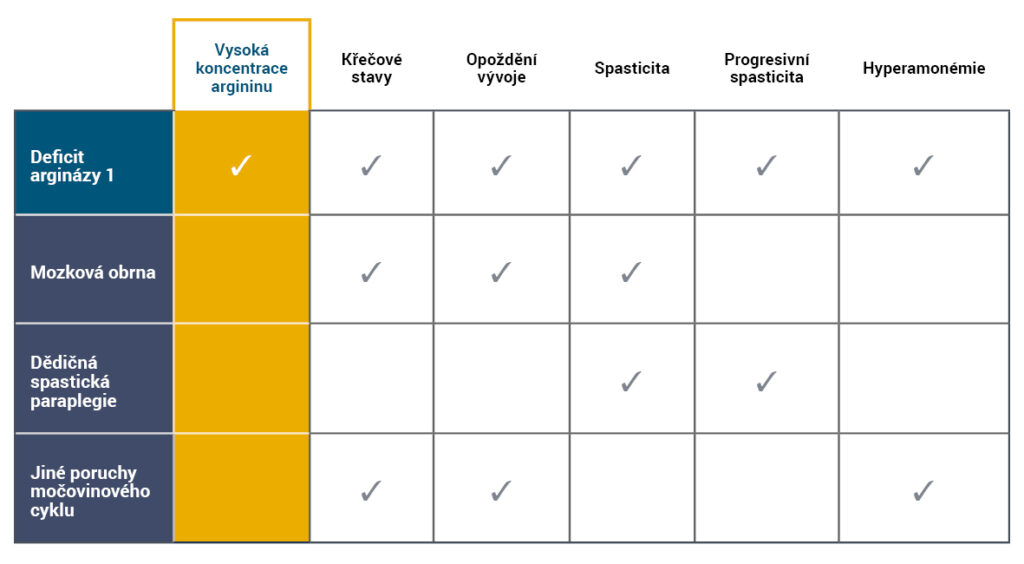
- Hyperamonémie není typickým znakem argininémie a k akutním hyperamonemickým atakám dochází zřídka4,8.
Novorozenecký screeningu má svoje limity, které mohou vést k falešně negativnímu nálezu, a to z řady důvodů:
- Hranice mezních hodnot argininu ve screeningu představuje problém, jelikož testování může ovlivnit přenos metabolitů, jako je arginin mezí krvi matky a dítěte9,10.
- Algoritmus screeningu a stanovené mezní hodnoty argininu se liší9.
- Ve většině evropských zemí není argininémie zahrnuta v novorozeneckém screeningu11. (V České repulice se screenuje od 2016).
Pozdě stanovená diagnóza spojená s pozdějším nástupem onemocnění vede k časné intervenci ve věku pouhých 6 let1.
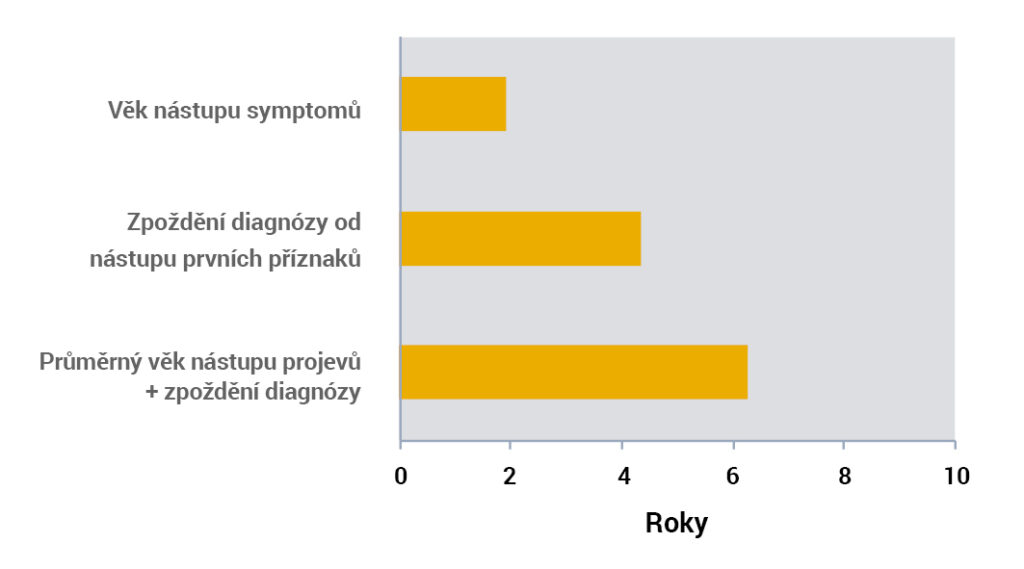
Argininémie může být zachycena v rámci analýzy hladin aminokyselin v krvi12,13.
Před laboratorním stanovením diagnózy je důležité správně vyhodnotit veškeré anamnestické údaje a provést dostatečné fyzikální vyšetření pacienta.
Ověřte zvýšenou koncentraci argininu, která je typickým projevem argininémie3,12,13.
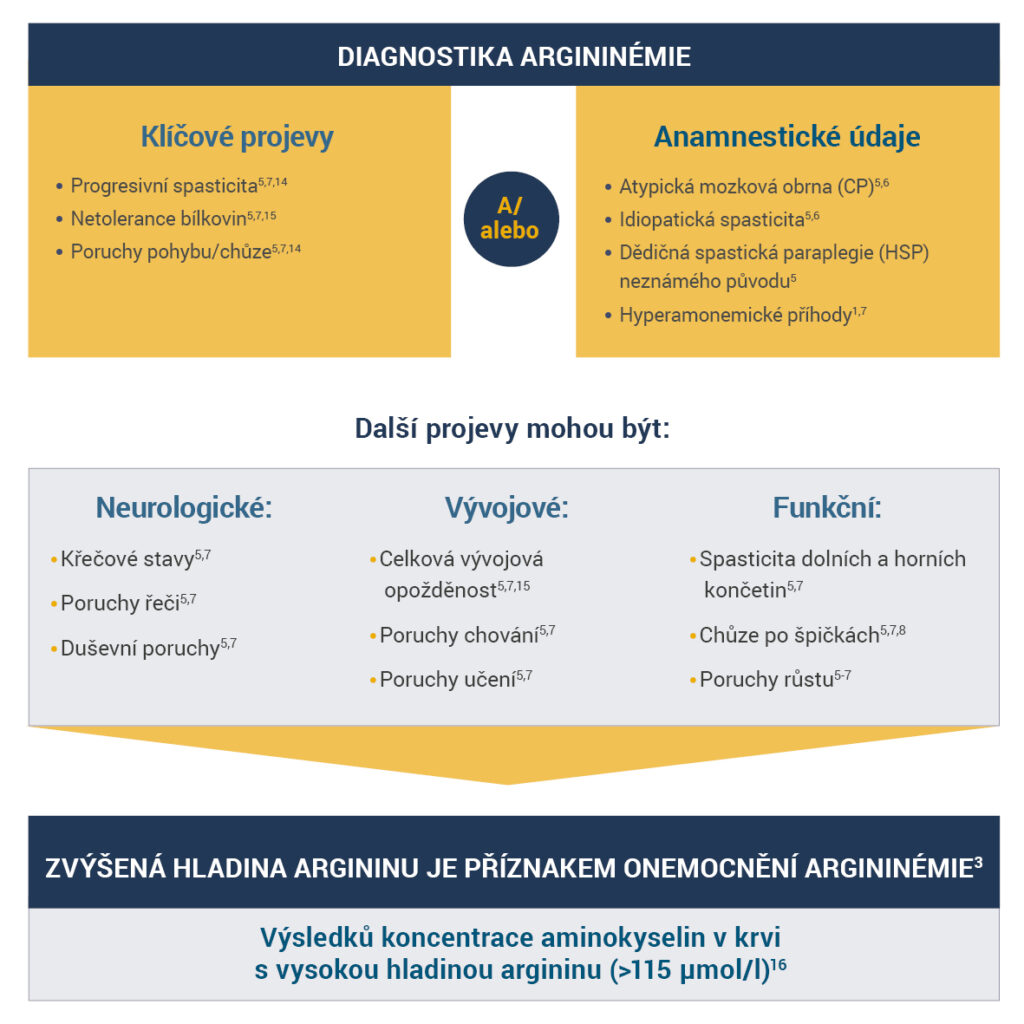

Je- li koncentrace argninu zvýšená, můžeme diagnózu argininémie potvrdit genetickým testováním†.
†Kvůli variabilitě genotypů ARG1 ještě nebyly identifikovány všechny mutace způsobující onemocnění.
Odkazy:
1. Huemer M, et al. J Inherit Metab Dis. 2016;39:331-340. 2. Edwards RL, et al. J Inherit Metab Dis. 2009;32:S197-S200. 3. De Deyn PP, et al. Hyperargininemia: a treatable inborn error of metabolism. In: Guanidino Compounds in Biology and Medicine. London, UK: John Libbey Company Ltd; 1997:53-69. 4. Burrage LC, et al. Hum Mol Genet. 2015;24:6417-6427. 5. Carvalho DR, et al. Pediatr Neurol. 2012;46:369-374. 6. Prasad A, et al. J Child Neurol. 1997;12:301-309. 7. Crombez EA, Cederbaum SD. Mol Genet Metab. 2005;84:243-251. 8. Scaglia F, Lee B. Am J Med Genet C Semin Med Genet. 2006;142C:113-120. 9. Therrell BL, et al. Mol Genet Metab. 2017;121:308–313. 10. Pitt JJ. Clin Biochem Rev. 2010;31:57-68. 11. Loeber JG, Platis D, Zetterström RH et al. Int J Neonatal Screening. 2021;7:15. 12. Sun A, et al. Arginase deficiency. In: Adam MP, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2020. 13. Ah Mew N, et al. Urea Cycle Disorders Overview. 2003. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1217/. Accessed November 26, 2021. 14. Cai X, et al. Medicine (Baltimore). 2018;97:e9880. 15. Bélanger SA, et al. Paediatr Child Health. 2018;23:403-410. 16. Lüneburg N, et al. J Nutr. 2011;141:2186-2190.